
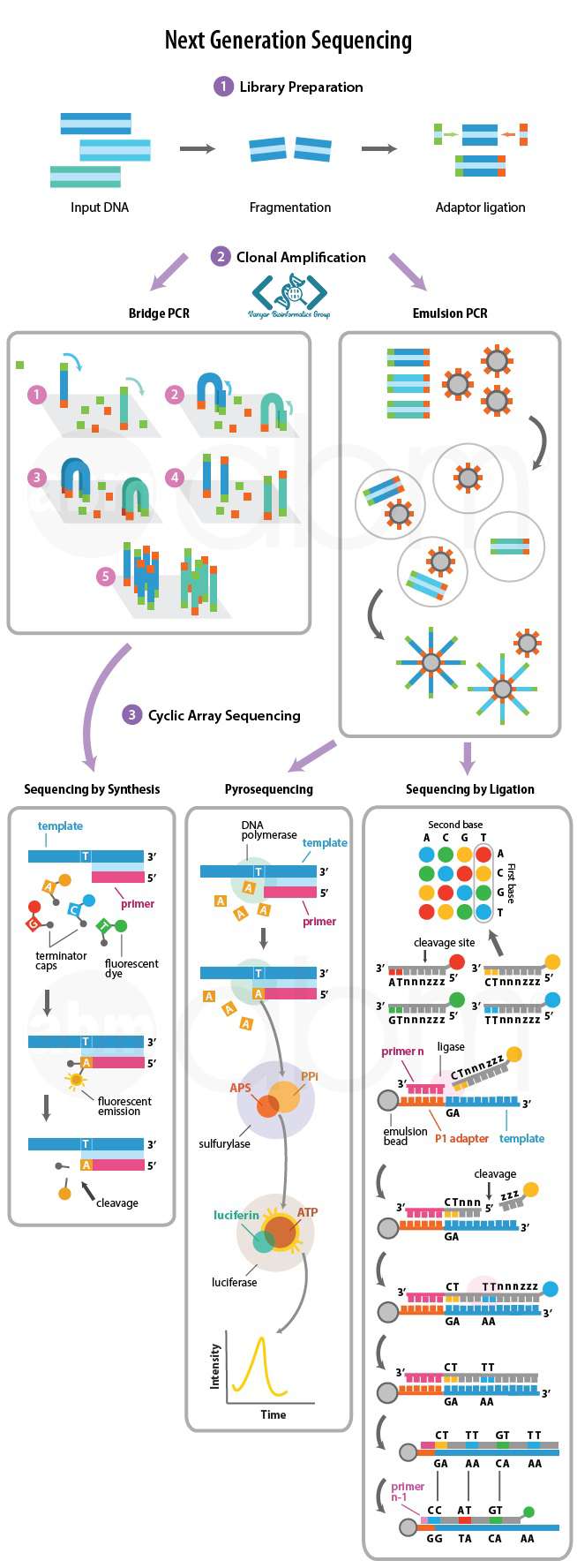
فرآیند اصلی توالی یابی نسل بعد (NGS) شامل توالی یابی کتابخانه های (library) حاصل از برش DNA و مونتاژ آنها برای تشکیل یک توالی ژنومی می باشد.
NGS از پلتفرم های مختلفی همچون توالی یابی با سنتز (SBS)، Pyrosequencing، و توالی یابی با لیگاسیون (SBL) برای تعیین سریع و دقیق توالی DNA استفاده می کند.
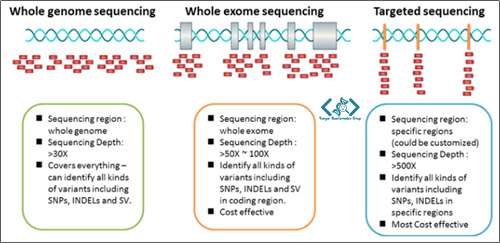
NGS می تواند برای تعیین توالی DNA هر موجود زنده ای استفاده شود و اطلاعات ارزشمندی را در پاسخ به تقریباً هر سؤال بیولوژیکی ارائه دهد. توالییابی DNA را میتوان برای مناطق کوچک و هدفمندی از ژنوم، و یا برای کل ژنوم به کار برد و محققان را قادر ساخت تا وضعیت سلامت و بیماری را بهتر درک کنند.
پیشنهاد: میزکار آنالیز نتایج توالی یابی سنگر امکان آنالیز توالی های مختلف را به سادگی و با چندکلیک فراهم کرده است. نتایج توسط کارشناسان بیوانفورماتیک وانیار گزارش و تایید می شوند. برای مشاهد پلن های کاربری به صفحه آنالیز نتایج توالی یابی سنگر وارد شوید: ورود به صفحه آنالیز نتایج توالی یابی سنگر.
در این مبحث با نگاهی اجمالی، به موضوعات زیر می پردازیم:
جامع ترین روش برای تجزیه و تحلیل ژنوم است.
کاهش سریع هزینه های توالی یابی و توانایی به دست آوردن اطلاعات ارزشمند در مورد کل کد ژنتیکی، این روش را به یک ابزار تحقیقاتی قدرتمند تبدیل کرده است.
با این نوع توالی یابی، زیرمجموعهای از ژنها یا مناطق خاصی از ژنوم، جداسازی و توالییابی میشوند.
این تکنیک به محققین اجازه میدهد تا زمان، هزینهها، و تجزیه و تحلیل را بر روی نواحی مورد نظر خود متمرکز کنند.
با استفاده از قدرت NGS، هم تجزیه و تحلیل گسترده ژنوم و هم رویکردهای هدفمند میتوانند بینشی در مورد الگوهای متیلاسیون DNA در سطح تک نوکلئوتید به محققان ارائه دهند.
از ادغام تکنیک کروماتین ایمونوپرسیپیتاسیون (ChIP) و توالی یابی، ابداع شده است.
این تکنیک برای شناسایی مکان های اتصال DNA به فاکتورهای رونویسی و سایر پروتئین ها در سطح ژنوم، روشی قدرتمند می باشد.
آمادهسازی کتابخانه برای پلتفرمهای اصلی NGS، مستلزم لیگاسیون یا اتصال الیگو آداپتورهای خاص به قطعاتی از DNA است که باید توالییابی شوند.
ابتدا، DNA به سایز بهینه ای که توسط پلتفرم مورد نظر تعیین شده، قطعه قطعه می شود.
چندین گزینه برای تکه تکه شدن DNA وجود دارد، از جمله روش های مکانیکی، آنزیمی و شیمیایی. روشهای مکانیکی ممکن است شامل برش هیدرودینامیکی توسط فراصوت، برش صوتی متمرکز، یا نبولیزاسیون باشد. همه روشهای مکانیکی پتانسیل ایجاد آسیب ناخواسته DNA را دارند، زیرا لزوماً باعث شکستگی نمیشوند!
آنزیم ها یک جایگزین مقیاس پذیر و مقرون به صرفه ارائه می دهند. واکنش های آنزیمی به طور کلی واکنش های کاملا ملایمی در مقایسه با برش مکانیکی هستند که تخریب نمونه ناخواسته یا آسیب DNA را به حداقل می رساند. آنزیم های مورد استفاده برای تکه تکه شدن DNA به سه نوع تقسیم می شوند: آنزیم های محدود کننده، آنزیم های ایجاد کننده شکاف (nicking enzymes) و ترانسپوزازها.
پیشنهاد: میزکار طراحی پرایمر وانیار امکان طراحی انواع مختلف پرایمرها را به سادگی و با چند کلیک فراهم کرده است. این پرایمرها تحت نظارت کارشناسان بیوانفورماتیک طراحی و اعتبارسنجی می شوند. برای مشاهد پلن های کاربری به صفحه طراحی پرایمر و پروب وارد شوید: ورود به صفحه طراحی پرایمر و پروب.
جهت افزودن آداپتورها به قطعات کتابخانه، به انتهای صاف (blunt) با دم تک نوکلئوتیدِ A در انتهای 3’ که قابلیت لیگاسیون دارد، نیاز است. از آنجایی که روش های برشی مکانیکی و آنزیمی تمایل به ایجاد انتهای آسیب دیده یا آزاد (overhang) دارند، بیشتر آنها قبل از اتصال آداپتورها نیاز به تعمیر دارند.
آنزیم ها کلید ترمیم نهایی هستند. یک مخلوط آنزیمی معمولی ممکن است، برای مثال، حاوی DNA پلیمراز T4 و پلی نوکلئوتید کیناز T4 (PNK) باشد. DNA پلیمراز T4 در حضور dNTP می تواند انتهای آزاد سر 5’ را پر کرده و انتهای آزاد سر 3’ را تا کوتاه کند تا انتهای صاف ایجاد شود. سپس T4 PNK می تواند نوکلئوتید انتهای 5’ را فسفریله کند.
مرحله ی بعد، A-tailing نیز به پلیمراز نیاز دارد. Taq DNA پلیمراز رایج ترین است زیرا دارای فعالیت ترانسفراز انتهایی است و به طور طبیعی یک آدنین انتهایی 3’ بر جای می گذارد. استفاده از هر یک این دست پلیمرازها انتهای A-tailed را ایجاد می کند که مکمل آداپتورهای توالی استاندارد هستند. افزودن آداپتور در این مرحله فقط نیاز به انکوباسیون با T4 DNA لیگاز دارد. این آنزیم هر دو انتهای صاف و به اصطلاح «چسبنده» را به هم میپیوندد.
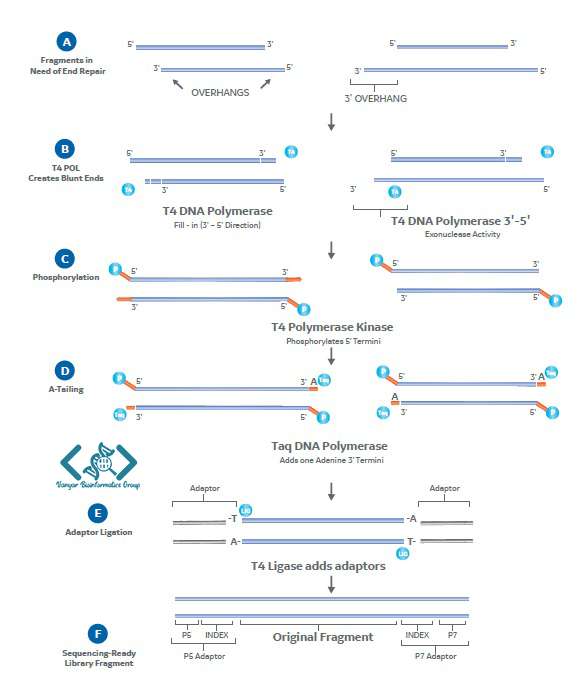
هر یک از این مراحل باید دقیق و کارآمد باشد تا کتابخانه بتواند داده های NGS قابل اعتماد تولید کند و بنابراین بر استفاده از آنزیم های با کیفیت بالا در شرایط واکنش بهینه تکیه دارد. نتیجه، یک کتابخانه از قطعات است که تقریبا برای توالی یابی آماده می باشد.









